Corporate European
Corporate European
 Denmark
Denmark Finland
Finland
 France
France
 Germany & Switzerland
Germany & Switzerland
 Greece
Greece Italy
Italy
 Latvia
Latvia
 Netherlands
Netherlands
 Norway
Norway
 Slovakia
Slovakia Spain & Portugal
Spain & Portugal
 Sweden
Sweden
 UK & Ireland
UK & Ireland
 Filtra
Filtra FHK Polska
FHK Polska PureMedion Kft.
PureMedion Kft. Elfa spol. s r. o.
Elfa spol. s r. o. Ecotip d.o.o.
Ecotip d.o.o.
Los retos a los que se enfrenta la industria farmacéutica son unos plazos de comercialización cortos y una productividad elevada, al tiempo que se cumplen las especificaciones y las normas de calidad, así como unos costes de producción razonables. En particular, la producción de medicamentos estériles está sujeta a requisitos especiales para minimizar los riesgos de contaminación microbiana y por partículas.
Existen estrictas directrices de la FDA y las GMP para limitar la exposición a dicha contaminación, evitando así daños graves o riesgos para la salud de los pacientes.
La esterilización por calor seco y la despirogenación se aplican para garantizar la esterilidad de los preparados farmacéuticos asépticos, tal como exige la normativa 21 CFR-211.94 de la FDA y el anexo 1 de las directrices GMP de la UE.
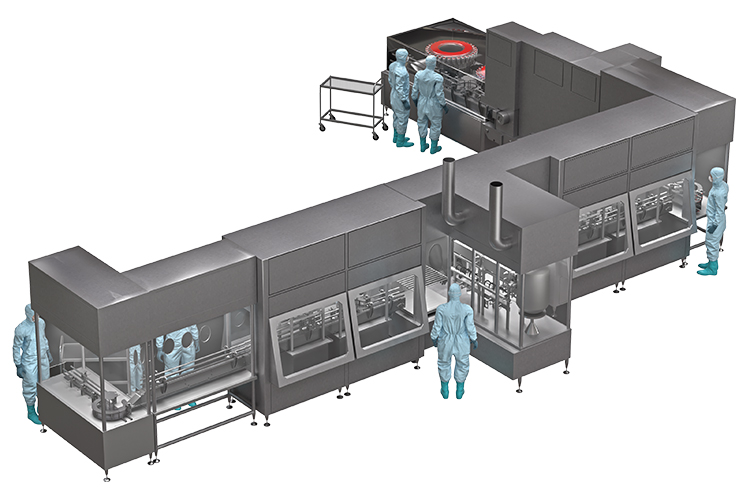
Envasado estéril. Una fase de producción crítica con una demanda creciente.
En el caso de los preparados asépticos, como viales, ampollas, cartuchos o jeringas precargadas, no es posible la esterilización terminal del envase final. Por lo tanto, el material de vidrio debe estar libre de contaminantes nocivos que puedan afectar al medicamento, antes del llenado. Dependiendo del proceso, se aplica la esterilización por calor seco o la despirogenación.
La esterilización suele aplicarse a una temperatura de 160-180 °C, para dejar el producto libre de microorganismos vivos. La despirogenación tiene por objeto eliminar o inactivar las endotoxinas, para lo que se requieren temperaturas más elevadas en la gama de 200-350 °C, y se lleva a cabo en hornos estáticos o en túneles para procesos automatizados y continuos.
Debido a la creciente demanda de envases estériles libres de pirógenos y de procesos rápidos, seguros y eficaces, la despirogenación en caliente es actualmente uno de los pasos más críticos en el proceso de fabricación de medicamentos estériles.